A high priority cure project is the development of stem cell gene therapy for Sickle Cell Disease (SCD) and a related condition, Beta-Thalassemia (BT), which together comprise of the hemoglobinopathies.
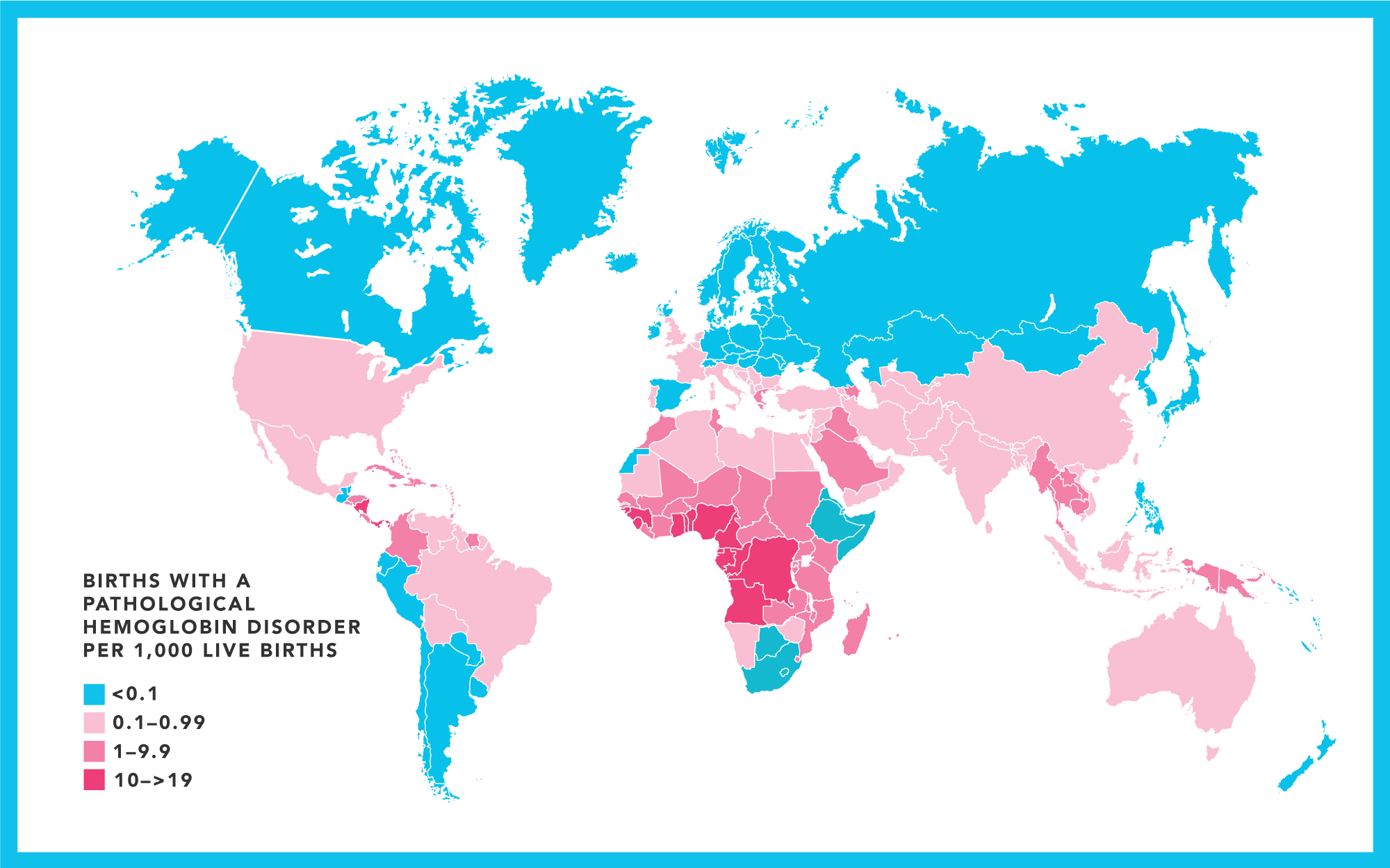
The hemoglobinopathies are the most prevalent monogenetic disorders in the world – over 5% of global population carry an abnormal hemoglobin gene. Between 300,000 –400,00 babies are born each year with a serious hemoglobin disorder.
Why we need a cure
Sickle cell disease (SCD) is a disorder of red blood cells with 20 million people suffering from the disease worldwide. SCD causes distressing acute-on-chronic morbidity, impaired health related quality of life (QoL), substantial healthcare utilization, and more than a two-decade reduction in life expectancy (www.nhlbi.nih.gov/science/cure-sickle-cell-initiative).
SCD disproportionately affects underserved populations, particularly in low- and middle- income countries (LMIC), including many in Africa. Allogeneic hematopoietic stem cell (HSC) transplantation offers the hope of cure for SCD but is severely limited by availability of HLA-matched donors as well as the costs and complexity of the treatment. Development of a onetime gene therapy using autologous hematopoietic stem cells (HSC) has the potential to dramatically recalibrate the healthcare environment and prognoses for SCD patients.
Beta Thalassemia (BT) similarly disproportionately affects underserved populations in the US and LMIC, including India, where 10,000 babies are born each year with the disease. The incidence of symptomatic cases is estimated to be approximately 1 in 100,000 individuals in the general population.
The disorder is particularly prevalent in the Mediterranean, Middle East, Africa, central Asia, the Indian subcontinent, and the Far East. Individuals in the US whose families are from these regions have a greater risk of carrying the genetic mutations that cause beta thalassemia. The effects of thalassemia disease on physical health can lead to physical deformity, growth retardation, and delayed puberty, which contributes to poor self-image for the patient. Accordingly, children with chronic physical illnesses such as thalassemia are prone to emotional and behavioral problems.
The chronicity and complications of thalassemia disease not only affect the QoL of patients but also hamper the entire family. Children with thalassemia in the pre-school and latency age groups are usually anxious and excessively dependent on their parents. At present, the complete and only treatment available for thalassemia disease major is bone marrow transplantation, which few patients in underserved communities can afford.
Studies to date
Several studies by different groups have shown clinical success using HSC gene therapy. Our collaborators at the National Institutes of Health have developed a HSC gene therapy that could be used to treat both SCD and BT. They have shown:
- Production of high-titer Lentiviral vectors expressing the β-globin gene
- Efficient lentiviral transduction and high-level engraftment of hematopoietic stem cells in humanized mice and non-human primates
- Clinically relevant levels of β-globin production in erythroid progenitors after transduction of SCD CD34+ stem cells.
Future Studies
A phase I/IIa clinical trial is planned for 2022. After initial safety and efficacy data at a single clinical center, a multi-center clinical trial is planned that is geographically dispersed throughout the US. After sufficient validation of safety and efficacy, clinical trials are planned for countries for which SCD and BT are highly prevalent and where there is high need for a cure.
Reference
Uchida N, Hsieh MM, Raines L, Haro-Mora JJ, Demirci S, Bonifacino AC, Krouse AE, Metzger ME, Donahue RE, Tisdale JF. Development of a forward-oriented therapeutic lentiviral vector for hemoglobin disorders. Nat Commun. 2019 Oct 2;10(1):4479. doi: 10.1038/s41467-019-12456-3. PMID: 31578323; PMCID: PMC6775231.
https://www.nature.com/articles/s41467-019-12456-3
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6775231/
Join our mission to help develop a cure for Sickle Cell Disease and Beta-Thalassemia Disease, and provide access to all communities.